Windkessel Redistribution and Transient Intracranial Hypertension After Cardiac Arrest: A Hemodynamic Hypothesis

The Paradox of Rising Intracranial Pressure After Cardiac Arrest
Over the years, I estimate I’ve spent close to two working (12-hour) days trying to find biomedical studies reporting intracranial pressure (ICP) after cardiac arrest. The paucity of experimental data about this crucial parameter is puzzling. Its absence in humans is more understandable, given that ICP monitoring is confined exclusively to neuroinjured patients and that medical interest, or at least clinical monitoring, of their ICP is nonexistent after death.
First-glance intuition suggests that ICP should rapidly decrease as circulatory pressure falls toward the mean circulatory filling pressure (MCFP), which is approximately 7–10 mm Hg (Guyton et al., 1954). And yet, animal studies have reported an acute increase in ICP within minutes of sudden cardiac arrest, in some cases approaching a doubling over baseline values (Figure 1) (Yashon et al., 1971; Park et al., 2010).
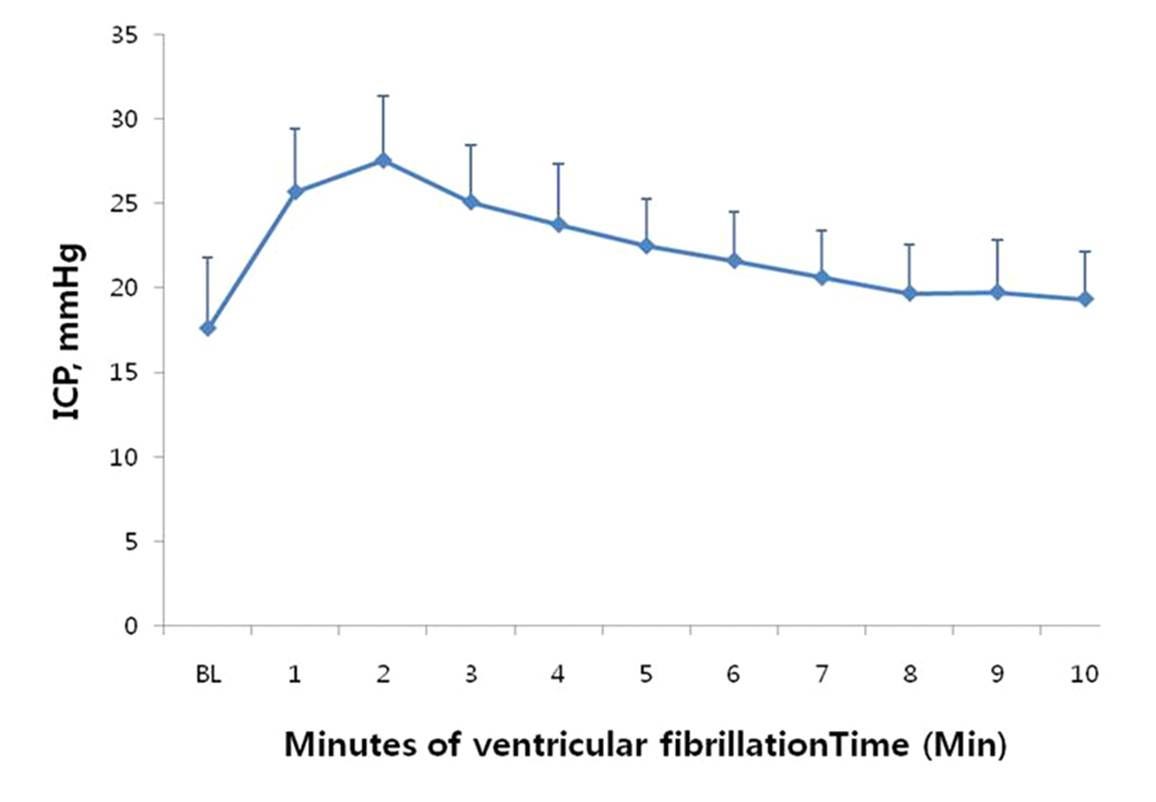
Figure 1. Increase in intracranial pressure in dogs subjected to sudden cardiac arrest (ventricular fibrillation), from Park et al (2010).
Why does this matter, particularly when the ICP slowly declines over the following 10 minutes or so (Figure 1)? The answer is that brain perfusion depends, in part, upon the cerebral perfusion pressure (CPP), classically defined as:
CPP = MAP − ICP
Where:
CPP = cerebral perfusion pressure
MAP = mean arterial pressure
ICP = intracranial pressure
This equation shows that cerebral blood flow depends on the pressure gradient that drives blood through the brain. As ICP rises, or MAP falls, CPP decreases. During cardiac arrest and CPR, the relationship between CPP and cerebral blood flow becomes highly nonlinear because autoregulation rapidly fails, and microvascular resistance, venous pressure, and vascular patency become the dominant determinants of flow (Branston et al., 1974; Guerci et al., 1985; Koehler et al., 1983; Paulson et al., 1990; Tweed et al., 1977). Nevertheless, CPP remains an important determinant of residual cerebral blood flow.
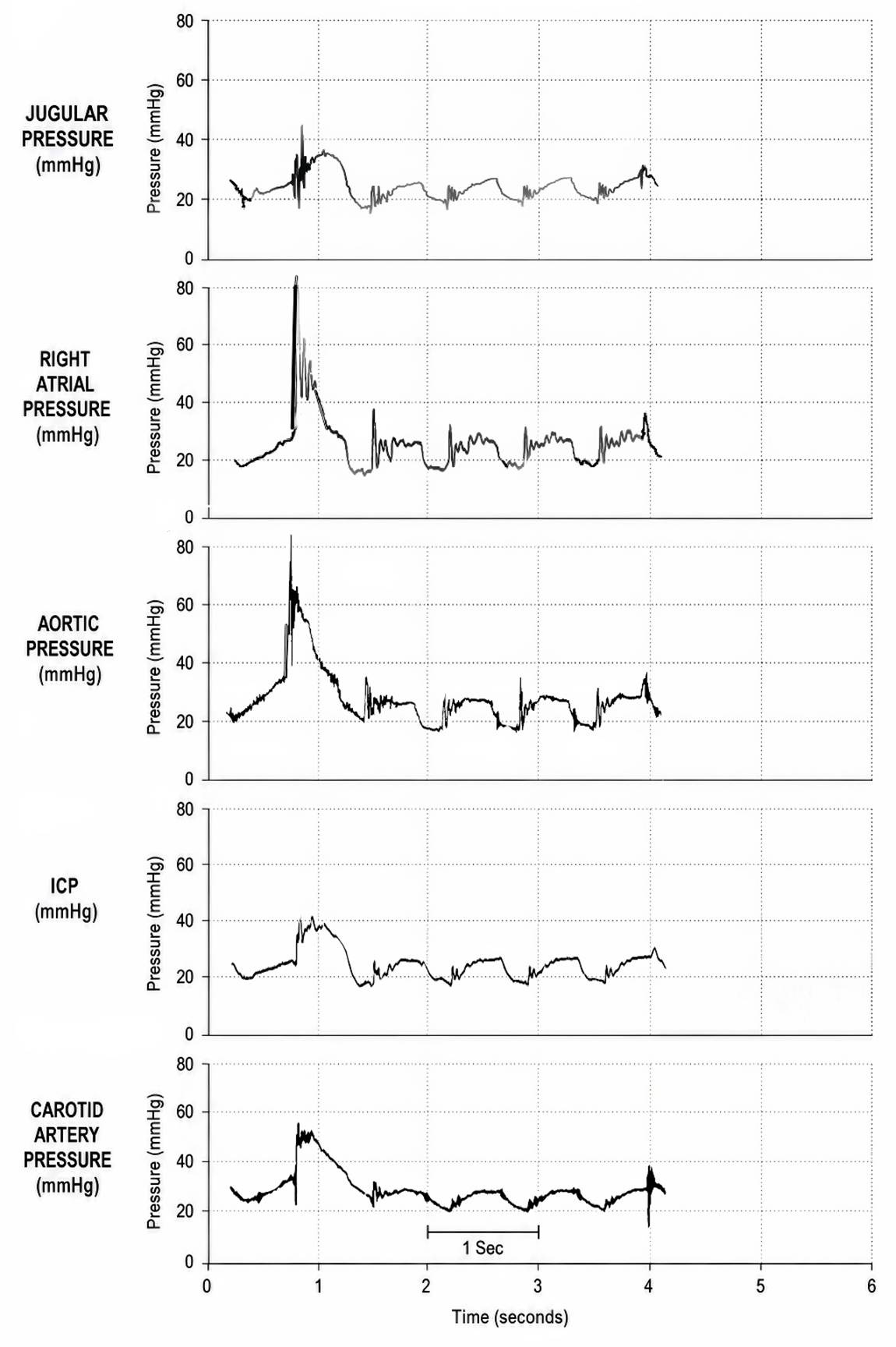
Figure 2. Hemodynamic pressure tracings during conventional closed-chest CPR in the dog, showing simultaneous increases in jugular venous pressure, right atrial pressure, aortic pressure, ICP, and carotid arterial pressure during chest compression. Note that the ICP averages 20 mm Hg or above—Redrawn from Koehler et al (1983).
This matters greatly in biostasis stabilization because conventional closed-chest CPR, even when expertly performed, typically produces substantially subphysiologic arterial pressures and cerebral blood flow (McDonald, 1981; Swenson et al., 1988; Jackson & Freeman, 1983). It is therefore concerning that closed-chest CPR can rapidly increase ICP from nominal values of approximately 5–15 mm Hg to 20–40 mm Hg (Figure 2) (Guerci et al., 1985; Koehler et al., 1983).
Thus, even relatively modest elevations of ICP will critically compromise cerebral perfusion during CPR. Indeed, microvascular flow may cease below a vessel-specific critical closing pressure (CrCP). In the cerebral circulation, effective or apparent critical closing pressure is typically 20–30 mm Hg under nominal adult conditions and may rise substantially with increased ICP, active vascular tone, hypocapnia, or intracranial hypertension. (Panerai, 2003; Calviello et al., 2023; Czosnyka et al., 1997). Consequently, reductions in CPP during low-flow states disproportionately impair capillary perfusion.
For example:
CPP = 50 − 30 = 20 mm Hg
Under such pressures, cerebral perfusion may become negligible.
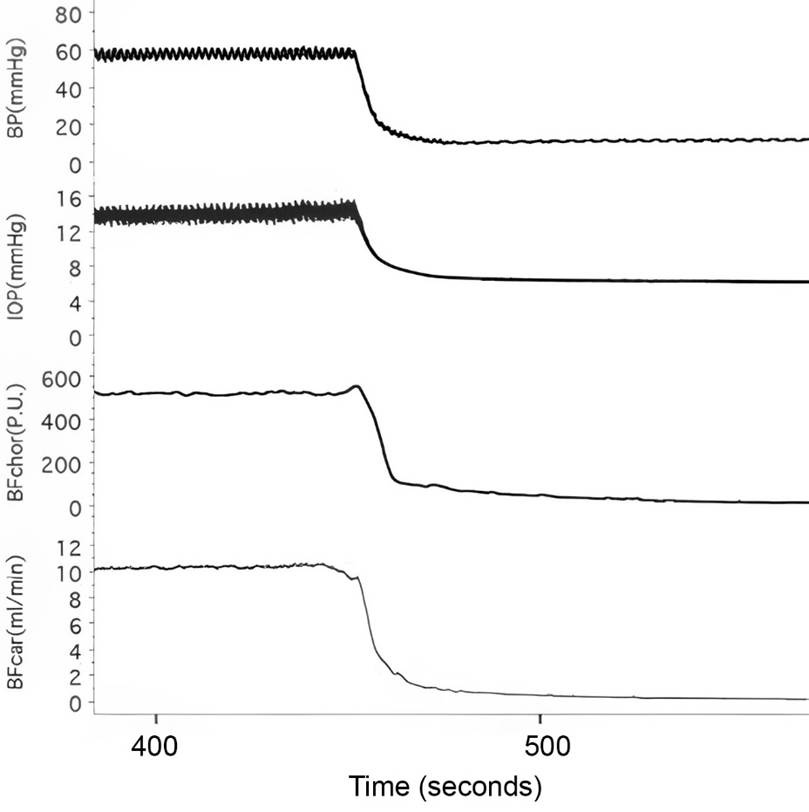
Figure 3. The decrease in intraocular pressure occurs immediately upon cardiac arrest induced with an overdose of pentobarbital in the deeply anesthetized rabbit. Continued venous outflow without corresponding arterial inflow results in a net loss of blood volume, which causes a fall in intraocular pressure (Kiel, 2010).
The increase in ICP following sudden cardiac arrest is made even more puzzling by the observation that intraocular pressure declines more or less in parallel with circulatory pressure following cardiac arrest. This comparison is intriguing because the retinal circulation shares important developmental and physiological features with the cerebral circulation (Kiel, 2010).
Windkessel Physiology and the Stressed Arterial Reservoir
I puzzled over these facts for far too long because I’ve known about “elastic reservoir action” since I first read Guyton’s Textbook of Medical Physiology decades ago (Guyton, 1981). Guyton explains that during ventricular systole, a portion of the stroke volume is transiently stored in the volume expansion of the aorta and large arteries. During diastole, this stored elastic strain energy is passively released, continuing to propel blood forward into the microcirculation even while the heart is resting. In the decades since I read Guyton’s sixth edition, the elastic recoil phenomenon has become known as the Windkessel effect.
“Windkessel” represents a conceptual and mathematical framework for understanding elastic reservoir behavior in terms of compliance and resistance. This ontological transition occurred because of the work of the German physiologist Otto Frank in 1899 (Westerhof, Lankhaar, Westerhof, 2009; Sagawa, Lie, & Schaefer, 1990). Frank formalized the arterial system as an “air-chamber” reservoir, analogous to the compressed-air chambers used in firefighting water pumps in the late 19th century (Figure 3). “Windkessel” is German for “air chamber” or “air vessel.” Frank mathematically demonstrated that, in combination, arterial compliance and peripheral resistance shape the arterial pressure waveform and maintain diastolic flow.
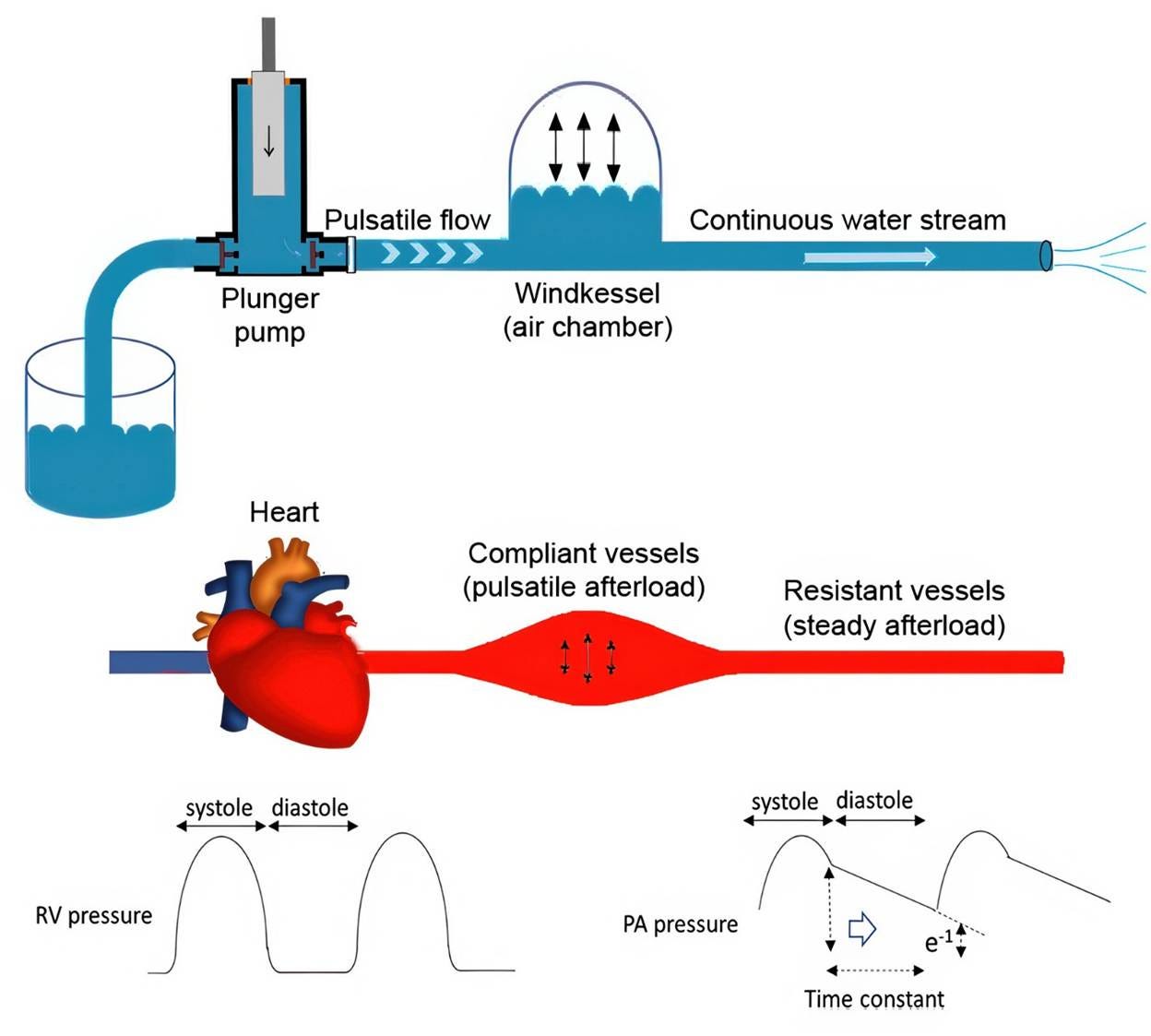
Figure 4. Windkessel model. The rhythmic water output from the plunger strokes is converted into a continuous downstream water jet via an air chamber. Physiologically, the plunger pump, air chamber, and downstream duct correspond to the heart, compliant arteries, and peripheral resistance vessels, respectively. Redrawn from Muneuchi et al., 2022.
In classical Windkessel modeling (Figure 4), the arterial circulation is represented as a single lumped elastic reservoir characterized by the total arterial compliance and peripheral resistance. The model strips away spatial vessel geometry and treats the entire arterial tree as a single compliant compartment capable of storing and releasing elastic energy.
Within this framework, the total arterial blood volume may be expressed as:
Vₐ(t) = V₀ₐ + Cₐ · Pₐ(t)
Where:
Vₐ(t) is the total arterial blood volume
V₀ₐ is the unstressed arterial volume
Cₐ is the total arterial compliance
Pₐ(t) is the instantaneous arterial pressure
The systemic arterial tree contains approximately 500–750 mL of blood at any given time. However, only ≈ 20% of this volume constitutes the stressed, pressure-generating reservoir available for rapid redistribution. Under nominal resting conditions, this stressed arterial volume is plausibly on the order of ~100–200 mL, based on systemic arterial compliance and mean arterial pressure, with the remainder representing relatively unstressed volume (Guyton et al., 1954; Guyton et al., 1973; Nichols et al., 2011; Sagawa et al., 1990; Westerhof et al., 2009)
During systole, cardiac inflow exceeds peripheral runoff, expanding the arterial reservoir and storing elastic strain energy in the arterial walls. During diastole, the heart ceases ejecting blood, and the stored elastic energy drives continued forward flow into the microcirculation:
dVₐ(t)/dt = Qᵢₙ(t) − Qₒᵤₜ(t)
At the moment of sudden cardiac arrest, cardiac inflow abruptly falls to zero while the stressed arterial volume remains pressurized. The Windkessel reservoir, therefore, undergoes passive discharge into the downstream venous capacitance system until arterial and venous pressures approach equilibrium near the mean circulatory filling pressure of 7-10 mm Hg.
The volume available for rapid redistribution following circulatory arrest is approximately:
ΔV = Cₐ · (Pₐ₀ − Pmcf)
Using physiologic values:
Cₐ ≈ 1–2 mL/mm Hg
Pₐ₀ ≈ 90 mm Hg
Pmcf ≈ 7–10 mm Hg
The predicted mobile reservoir volume is roughly 100–200 mL.
Windkessel Discharge as a Mechanism of Post-Arrest ICP Elevation
This observation has potentially important implications for intracranial pressure in cardiac arrest. According to the Monro-Kellie doctrine, the total volume of the intracranial contents (blood, cerebrospinal fluid, and brain tissue) must remain constant because they are encased within a rigid, non-compliant container (Mokri, 2001). Consequently, even a modest translocation of the stressed arterial reservoir into the intracranial vascular compartment—without a simultaneous and equal egress of venous blood or CSF—will lead to an immediate and disproportionate rise in ICP.”
Why Cytotoxic Edema Is Unlikely to Explain the Early ICP Rise
Because anoxic-ischemic depolarization (AID) occurs shortly after circulatory arrest, it was investigated as a possible cause of the post-arrest increase in ICP. However, cytotoxic edema cannot reasonably explain the early, stereotypical ICP rise beginning approximately 1.5 minutes after sudden cardiac arrest, because the large ionic movements required to generate substantial Gibbs free energy release water shifts are initiated by, or become dominant after, AID or terminal spreading depolarization (Dreier et al., 2018; Hansen, 1985; Somjen, 2001). In both experimental and human intracranial electocortical recordings, AID follows an initial period of electrical silence and generally occurs only after several minutes of severe ischemia.
Electrical activity in the cortex is suppressed within seconds of complete ischemia, whereas terminal depolarization typically occurs only after continued failure of perfusion and substrate delivery (Dreier et al., 2018; Graf et al., 2002; Hossmann & Zimmermann, 1974). In the human intracranial recordings reported by Dreier et al. (2018), terminal spreading depolarization occurred at a median of 3.9 minutes after the onset of the final decline in perfusion, with an interquartile range of 2.6–6.3 minutes. Therefore, an ICP rise beginning approximately 90 seconds after sudden cardiac arrest (Figure 1) is temporally difficult to reconcile with cytotoxic edema secondary to AID and is more consistent with rapid intravascular volume redistribution, such as Windkessel-mediated arterial-to-venous translocation of stressed arterial volume.
Intrathoracic pressure transmission occurs rapidly during CPR (Figure 2). However, it does not explain the increase in ICP during untreated arrest, in which intrathoracic pressure does not rise abruptly, and central venous pressure falls toward the mean circulatory filling pressure. Loss of autoregulatory vasoconstriction may increase vascular volume, but in the absence of ongoing cardiac output, it cannot by itself add substantial intracranial blood volume.
The most parsimonious explanation is therefore rapid intravascular redistribution: residual elastic recoil of the arterial Windkessel continues to propel blood into the cerebral vascular bed after ventricular ejection ceases, while venous outflow and intracranial compliance are insufficient to buffer the transient volume shift.
The brain within the rigid cranial vault is a low-compliance compartment. Consequently, redistribution of even a relatively small amount of blood volume may produce significant increases in ICP over a time course that is critically important for cerebral reperfusion during CPR.
Human Observations During the Dying Process
This interpretation led me to a further prediction: patients dying slowly in profound shock might exhibit little or no post-arrest increase in ICP. In such patients, arterial pressure typically declines progressively before arrest, reducing the stressed arterial reservoir available for rapid Windkessel discharge. Simultaneously, reduced blood volume and increased venous capacitance may permit greater buffering of any residual redistribution. Most slowly dying patients experience dramatically reduced or absent fluid intake in the days and hours preceding cardiac arrest, with a likely consequent reduction in venous capacitance. Under this hypothesis, only individuals with nominal hemodynamics, such as otherwise healthy individuals who experience sudden cardiac arrest, would experience a post-arrest surge in ICP.
With the advent of sophisticated, easily accessible AI systems, I was able to test this hypothesis mathematically. More importantly, I could cross-check calculations across multiple systems, even if I could not independently derive every step.
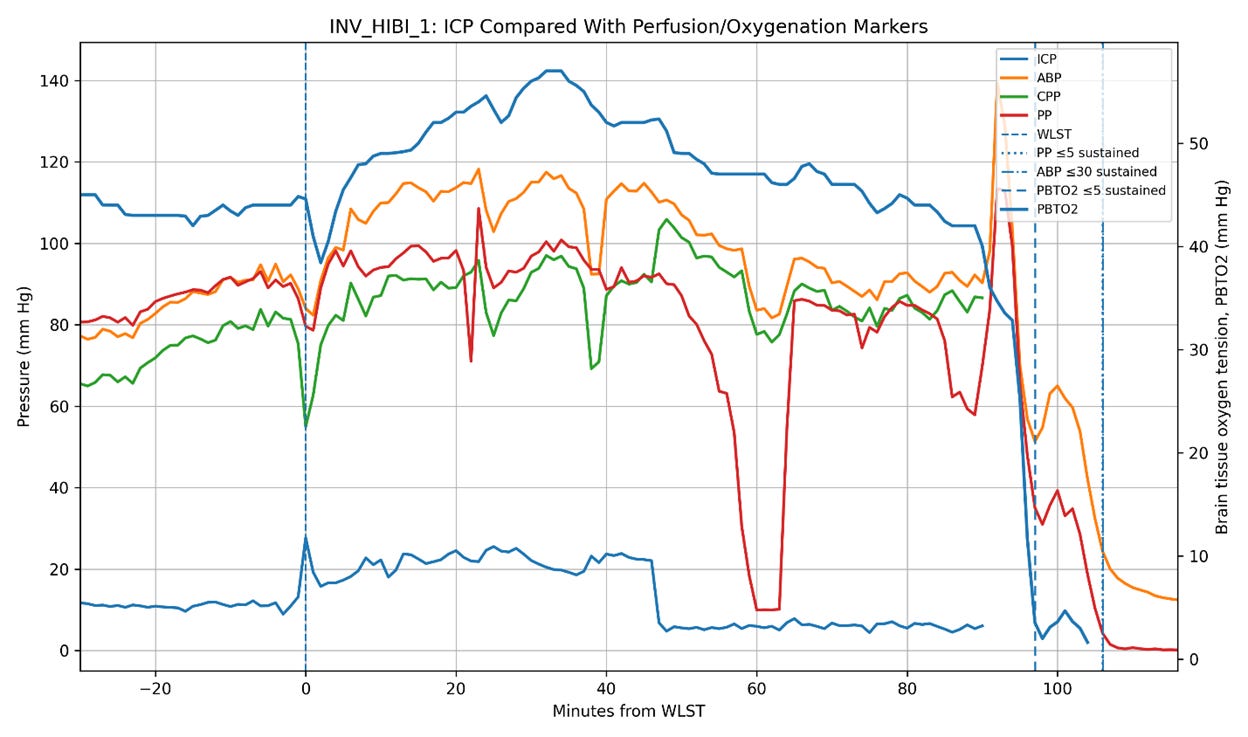
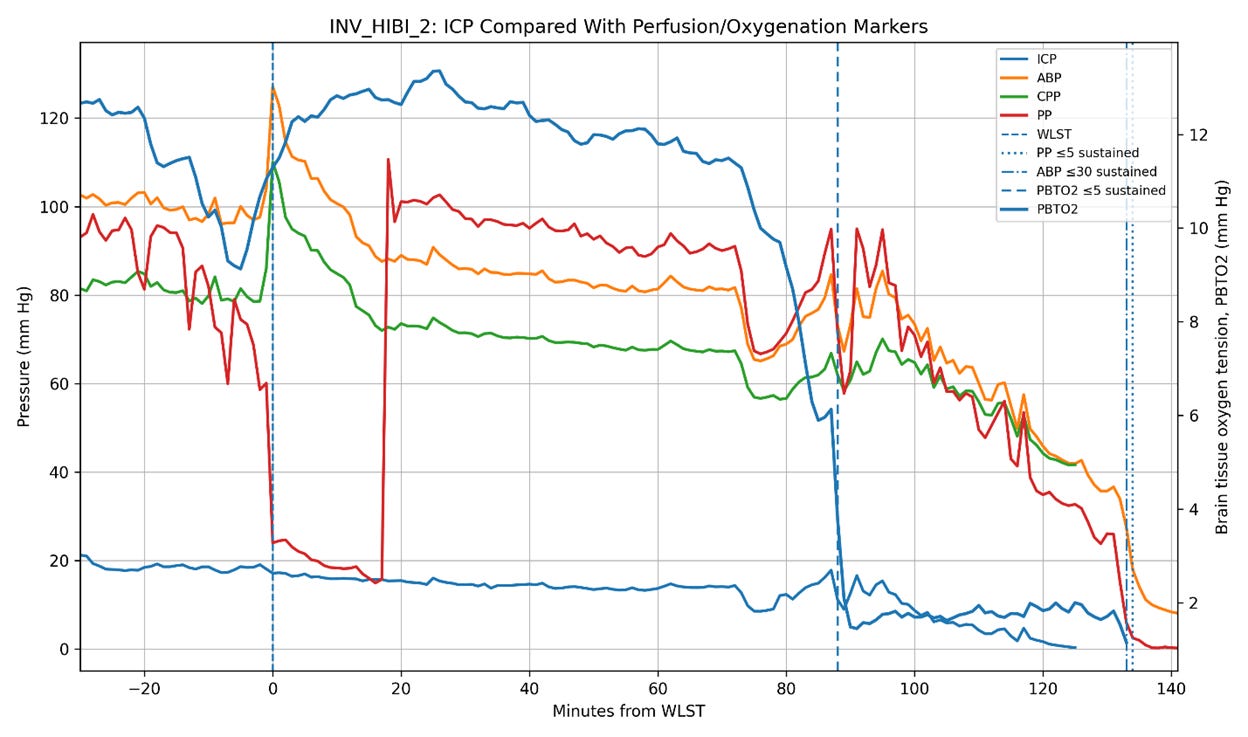
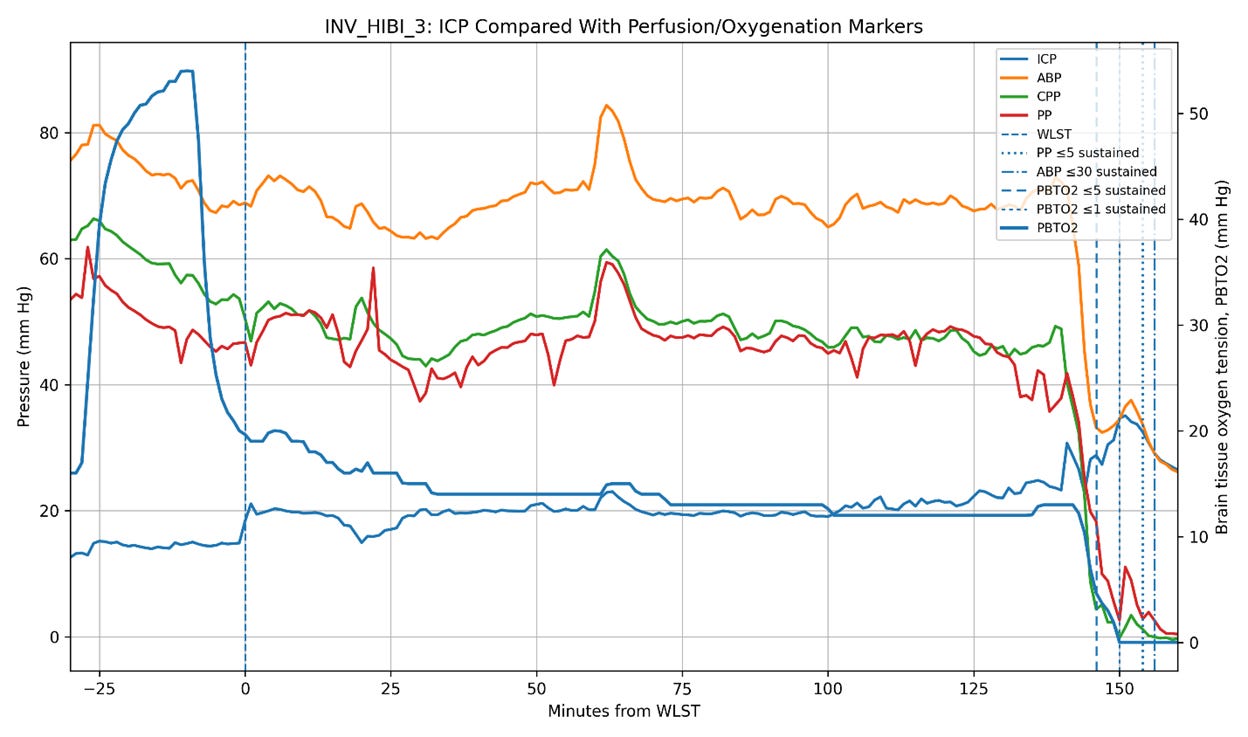
Figure 5. Intracranial pressure (ICP), arterial blood pressure (ABP), cerebral perfusion pressure (CPP), pulse pressure (PP), and brain tissue oxygen tension (PbtO2) trajectories in the three Bird et al. patients undergoing invasive neuromonitoring during withdrawal of life-sustaining therapy (WLST). Time zero corresponds to WLST. Vertical dashed lines indicate sustained collapse of pulse pressure (PP ≤ 5 mm Hg), sustained severe hypotension (ABP ≤ 30 mm Hg), and severe cerebral hypoxia (PbtO2 ≤ 5 mm Hg). In two patients (INV_HIBI_1 and INV_HIBI_2), ICP progressively declined in parallel with systemic circulatory collapse, reaching approximately 6 mm Hg and 0.3 mm Hg, respectively, near the time of pulse-pressure collapse. In contrast, INV_HIBI_3 exhibited persistent moderate intracranial hypertension throughout the agonal period, with ICP remaining approximately 20–35 mm Hg even as systemic perfusion pressures and PbtO2 progressively declined. Notably, severe cerebral hypoxia preceded pulse-pressure collapse in all three patients, indicating substantial impairment of cerebral perfusion before terminal circulatory arrest. These observations suggest that post-arrest ICP behavior is heterogeneous and may depend on the residual stressed arterial reservoir volume, the systemic hemodynamic state, and the intracranial compliance present at the time circulation ceases.
Conclusion
Of course, hypotheses remain hypotheses until tested against empirical observation. That is why the publication of the paper by Bird et al. (2025) documenting the physiology of dying in humans was so interesting. In that study, three patients were monitored for ICP during the dying process. Two of the three exhibited post-arrest declines in ICP broadly consonant with the decline in circulatory pressures, while the third demonstrated an increase in ICP following circulatory arrest (Figure 5).
These observations are at least consistent with the possibility that the magnitude of post-arrest ICP elevation depends upon the residual stressed arterial volume present at the time circulation ceases. In otherwise healthy, normovolemic animals subjected to sudden cardiac arrest, rapid Windkessel discharge may transiently increase ICP over a time course critically important to cerebral reperfusion. In profoundly shocked or hypovolemic patients, the effect may be substantially blunted.
The available evidence remains sparse, and the observations from three severely neurologically injured patients are obviously insufficient to establish a general physiological principle. Nevertheless, the findings are sufficiently consistent with classical Windkessel dynamics to warrant publication and further investigation.
References
Bird, J. D., Hornby, L., Hirsch-Reinshagen, V., et al. (2025). Characterizing the physiology of circulatory arrest in humans. Nature Medicine, 31, 3542–3552. https://doi.org/10.1038/s41591-025-03889-z
Branston, N. M., Symon, L., Crockard, H. A., & Pasztor, E. (1974). Relationship between the cortical evoked potential and local cortical blood flow following acute middle cerebral artery occlusion in the baboon. Experimental Neurology, 45(2), 195–208. https://doi.org/10.1016/0014-4886(74)90112-5
Calviello, L., Donnelly, J., Cardim, D., Robba, C., Zeiler, F. A., Smielewski, P., Czosnyka, M., & Aries, M. J. H. (2023). Cerebral critical closing pressure in neurocritical care: A scoping review. Journal of Cerebral Blood Flow & Metabolism, 43(7), 1077–1092. https://doi.org/10.1177/0271678X231172854
Czosnyka, M., Smielewski, P., Kirkpatrick, P., Piechnik, S., Laing, R., & Pickard, J. D. (1997). Critical closing pressure in cerebrovascular circulation. Journal of Neurology, Neurosurgery & Psychiatry, 62(3), 240–246.
Guerci, A. D., Shi, A. Y., Levin, H., Tsitlik, J., Weisfeldt, M. L., & Chandra, N. (1985). Transmission of intrathoracic pressure to the intracranial space during cardiopulmonary resuscitation in dogs. Circulation Research, 56(1), 20–30. https://doi.org/10.1161/01.RES.56.1.20
Guyton, A. C. (1981). Textbook of medical physiology (6th ed.). W.B. Saunders Company.
Guyton, A. C., Jones, C. E., & Coleman, T. G. (1973). Circulatory physiology: Cardiac output and its regulation. W.B. Saunders.
Guyton, A. C., Polizo, D., & Armstrong, G. G. (1954). Mean circulatory filling pressure measured immediately after cessation of heart pumping. American Journal of Physiology, 179, 261–267. https://doi.org/10.1152/ajplegacy.1954.179.2.261
Jackson, R. E., & Freeman, S. B. (1983). Hemodynamics of cardiac massage. Emergency Medicine Clinics of North America, 1(3), 501–513. PMID: 6396069
Kiel, J. W. (2010). The ocular circulation. Morgan & Claypool Life Sciences.
Koehler, R. C., Chandra, N., Guerci, A. D., Tsitlik, J., Traystman, R. J., Rogers, M. C., & Weisfeldt, M. L. (1983). Augmentation of cerebral perfusion by simultaneous chest compression and lung inflation with abdominal binding after cardiac arrest in dogs. Circulation, 67(2), 266–275. https://doi.org/10.1161/01.CIR.67.2.266
McDonald, J. L. (1981). Systolic and mean arterial pressures during manual and mechanical CPR in humans. Critical Care Medicine, 9(5), 382–383. https://doi.org/10.1097/00003246-198105000-00015
Mokri B. (2001). The Monro-Kellie hypothesis: applications in CSF volume-pressure relationships. Mayo Clinic Proceedings, 76(6), 641–644.
Muneuchi, J., Ezaki, H., Sugitani, Y., & Watanabe, M. (2022). Comprehensive assessments of pulmonary circulation in children with pulmonary hypertension associated with congenital heart disease. Frontiers in Pediatrics, 10, 1011631. https://doi.org/10.3389/fped.2022.1011631
Nichols, W. W., O’Rourke, M. F., & Vlachopoulos, C. (2011). McDonald’s blood flow in arteries: Theoretical, experimental and clinical principles (6th ed.). Hodder Arnold.
Panerai, R. B. (2003). The critical closing pressure of the cerebral circulation. Medical Engineering & Physics, 25(8), 621–632. https://doi.org/10.1016/S1350-4533(03)00065-1
Park, J., Ristagno, G., Chung, S., Weng, Y., Wu, X., Sun, S., Weil, M. H., & Tang, W. (2010). The potential mechanism of sudden increase in intracranial pressure during initial cardiac arrest. Circulation, 122(Suppl. 21), A45.
Paulson, O. B., Strandgaard, S., & Edvinsson, L. (1990). Cerebral autoregulation. Cerebrovascular and Brain Metabolism Reviews, 2(2), 161–192. PMID: 2201348.
Sagawa, K., Lie, R. K., & Schaefer, J. (1990). Translation of Otto Frank’s paper “Die Grundform des Arteriellen Pulses” Zeitschrift für Biologie, 37, 483–526 (1899). Journal of Molecular and Cellular Cardiology, 22(3), 253–254. https://doi.org/10.1016/0022-2828(90)91459-K
Swenson, R. D., Weaver, W. D., Niskanen, R. A., Martin, J., & Dahlberg, S. (1988). Hemodynamics in humans during conventional and experimental methods of cardiopulmonary resuscitation. Circulation, 78(3), 630–639. https://doi.org/10.1161/01.cir.78.3.630
Tweed, W. A., Wade, J. G., & Davidson, W. J. (1977). Mechanisms of the “low-flow” state during resuscitation of the totally ischemic brain. Canadian Journal of Neurological Sciences, 4(1), 19–23. PMID: 837260.
Westerhof, N., Lankhaar, J. W., & Westerhof, B. E. (2009). The arterial Windkessel. Medical & Biological Engineering & Computing, 47(2), 131–141. https://doi.org/10.1007/s11517-008-0359-2
Yashon, D., Wagner, F. C., Jr., White, R. J., Albin, M. S., & Locke, G. E. (1971). Intracranial pressure during circulatory arrest. Brain Research, 31(1), 139–150. https://doi.org/10.1016/0006-8993(71)90638-X